Note
Data used throughout this example can be found in <ROOT>/examples/contaminant_example. If SIMBAD is part of your CCP4 installation,
then the example files can be downloaded as part of the GitHub repository.
Check out this page explaining the simbad-contaminant script command line options.
In this example, the simbad-contaminant script simply takes the crystallographic data file in MTZ format, and runs the contaminant search on your local machine.
simbad-contaminant \
-nproc 4 \
-organism CHICK \
Upon running SIMBAD results will be output to the terminal:
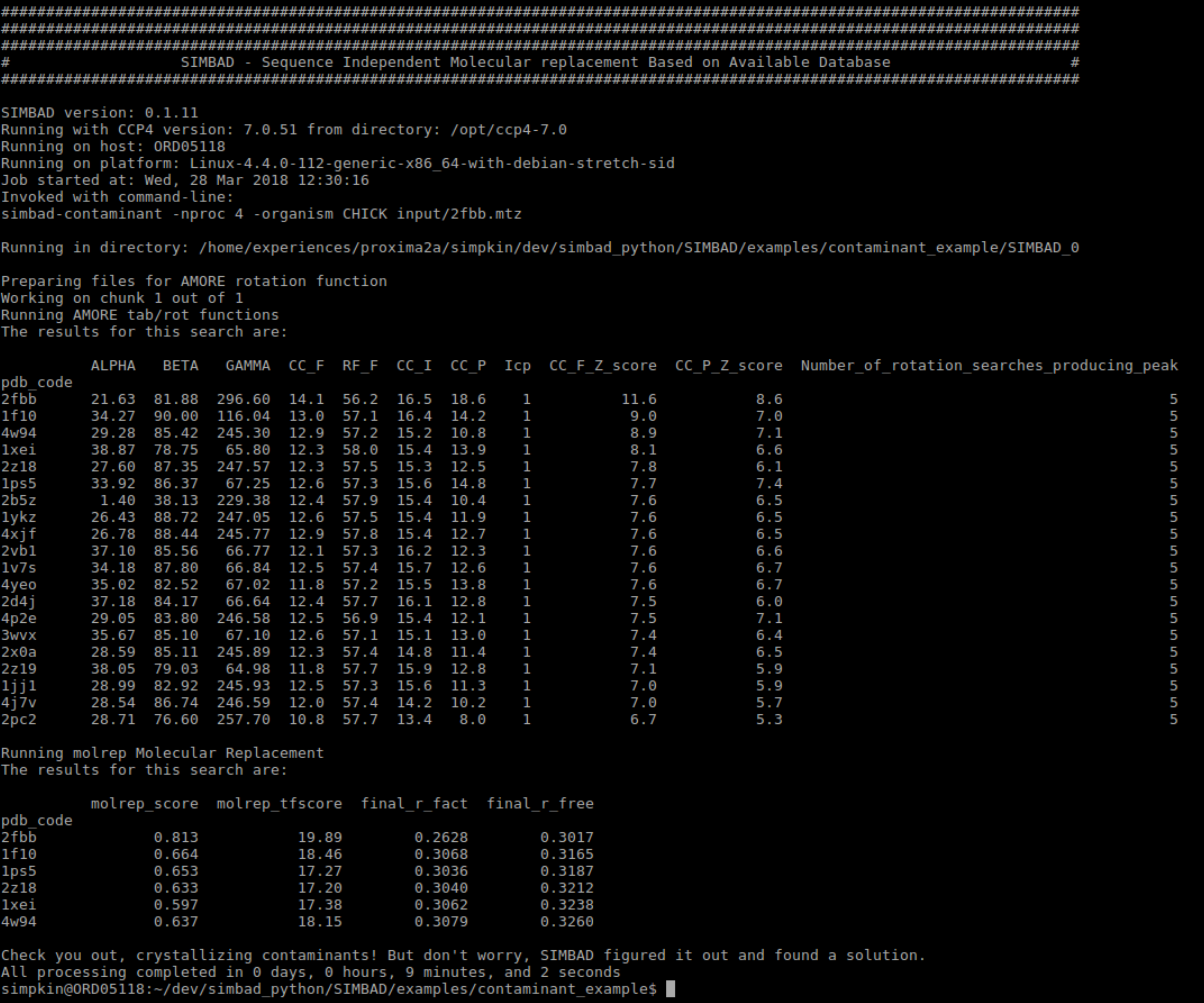
The Contaminant Search outputs 2 tables. Below you can find information about each:
This shows the results from the AMORE Rotation Search carried out on the contaminant database. The columns of the table are:
The structures are scored by CC_F_Z_score score where a higher score is better.
Molecular replacement is performed on the top 20 structures identified by the contaminant database AMORE Rotation search. This section displays the results of that molecular replacement.
By default SIMBAD runs Molecular replacement using MOLREP. If run the following columns are added to the table:
Alternatively SIMBAD can run Molecular replacement using PHASER. If run the following columns are added to the table:
Following Molecular replacement, refinement is run using REFMAC. This add the following columns are added to the table:
Note
Typically a result with a final_r_fact and a final_r_free below 0.45 is indicative of a solution.
Additionally if there is anomalous signal in your dataset SIMBAD will try to validate the quality of the molecular replacement solution using by plotting the peaks from a phased anomalous fourier map. If run the following columns are added to the table: